(array([[1.4758799 , 1.42249684, 1.51688992],
[1.1620039 , 1.04449795, 1.43987427],
[0.67112498, 0.91171351, 0.92751922]]),
array([[1.4758799 , 1.42249684, 1.51688992],
[1.1620039 , 1.04449795, 1.43987427],
[0.67112498, 0.91171351, 0.92751922]]))Linear algebra
Numerical linear algebra is a huge topic
We’ll look at how common operations are performed in NumPy and SciPy, and some applications in physics
Linear algebra with NumPy
Multiplying matrices is easy in NumPy using
np.matmul@operator gives shortcut
- You’ll get an error if your matrices don’t match…
--------------------------------------------------------------------------- ValueError Traceback (most recent call last) Cell In[2], line 3 1 C = np.random.rand(2, 3) 2 D = np.random.rand(4, 2) ----> 3 C @ D ValueError: matmul: Input operand 1 has a mismatch in its core dimension 0, with gufunc signature (n?,k),(k,m?)->(n?,m?) (size 4 is different from 3)
If either
AorBhas rank greater than two: treat as a stack of matrices, with each matrix in last two indicesUsual broadcasting rules then apply to remaining indices
- Most versatile is
np.einsum: explicitly translate expressions using the Einstein summation convention
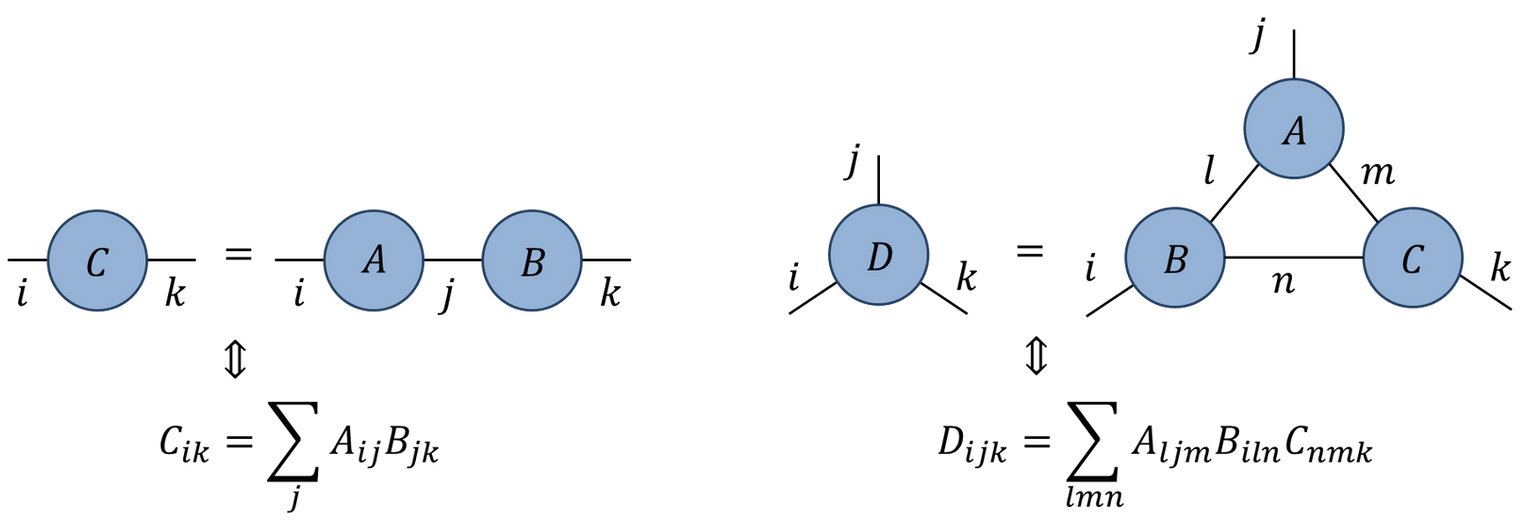
\[ \left[A\cdot B\right]_{ik} = \sum_{j} A_{ij}B_{jk} = A_{ij}B_{jk} \]
array([[1.4758799 , 1.42249684, 1.51688992],
[1.1620039 , 1.04449795, 1.43987427],
[0.67112498, 0.91171351, 0.92751922]])\[ \operatorname{tr}\left[A\cdot B\right] = \sum_{i,j} A_{ij}B_{ji} = A_{ij}B_{ji} \]
In what order should multiple contractions be evaluated?
How should loops be nested?
Recall: should evaluate \(M_1 M_2\cdots M_n \mathbf{v}\) as \(O(N^2)\) matrix-vector multiplications, rather than \(O(N^3)\) matrix-matrix multiplications followed by matrix-vector multiplication
In general no efficient algorithm to find best way!
einsumcan use a “greedy” algorithm (contracting pair of tensors with lowest cost at each step)Information on contraction order provided by
np.einsum_path
- Inversion (
np.linalg.inv) - Calculation of determinant (
np.linalg.det) - Eigenvalues and eigenvectors (
np.linalg.eigornp.linalg.eighfor hermitian problems)
…inherit their complexity from \(O(N^3)\) complexity of matrix multiplication
Power method
Better methods available if only want largest (or smallest) eigenvalue and eigenvector (e.g. QM ground state)
Simplest is Power method
- Start from arbitrary vector \(\mathbf{b}_0\)
- Multiply repeatedly by matrix \(A\)
- Result tends to dominant eigenvector (largest magnitude eigenvalue)
- Convenient to normalize each time
\[ \mathbf{b}_{k+1} = \frac{A \mathbf{b}_k}{\lVert A\mathbf{b}_k\rVert} \]
\[ \lim_{k\to\infty}\mathbf{b}_k = \mathbf{v}_\text{dom} \]
\[ A\mathbf{v}_\text{dom} = \lambda_\text{dom}\mathbf{v}_\text{dom} \]
Already met this idea when we discussed Markov chains
Relevant matrix was \(\mathsf{P}_{jk}=p(j|k)\geq 0\) of transition probabilities, which is stochastic:
\[ \sum_j \mathsf{P}_{jk} = 1 \]
- Guarantees that dominant eigenvalue is one and dominant eigenvector has interpretation of the stationary distribution
PageRank
- Google’s PageRank algorithm assesses relative importance of webpages based on structure of links between them

PageRank imagines a web crawler navigating between pages according to a transition matrix \(\mathsf{P}\)
Stationary distribution \(\boldsymbol{\pi}\) satisfying\[ \mathsf{P}\boldsymbol{\pi} = \boldsymbol{\pi} \] then interpreted as a ranking
Page \(j\) more important than page \(k\) if \(\boldsymbol{\pi}_j>\boldsymbol{\pi}_k\)
- Problem if Markov chain is non-ergodic: state space breaks up into several independent components, leading to nonunique \(\boldsymbol{\pi}\)
\[ \begin{equation} \mathsf{P}=\begin{pmatrix} 0 & 1 & 0 & 0 \\ 1 & 0 & 0 & 0 \\ 0 & 0 & 0 & 1 \\ 0 & 0 & 1 & 0 \end{pmatrix} \end{equation} \]
- First two pages and last two do not link to each other, so \(\boldsymbol{\pi}^T = \begin{pmatrix} \pi_1 & \pi_1 & \pi_2 & \pi_2 \end{pmatrix}\) is a stationary state for any \(\pi_{1,2}\)
Way out is to modify Markov chain to restore ergodicity and give a unique \(\boldsymbol{\pi}\)
Crawler either moves as before with probability \(\alpha\) or moves with probability \(1-\alpha\) to a random webpage \[ \alpha\mathsf{P} + (1-\alpha)\mathbf{t} \mathbf{e}^T \] where \(\mathbf{e}^T= (1, 1, \ldots 1)\) and \(\mathbf{t}\) is a “teleporting” vector
Matrix has positive (i.e. \(>0\)) entries: there is a unique stationary state (and hence ranking)
Further modification is required to teleport away from “dangling” webpages without any outgoing links
Power method is basis of more sophisticated algorithms such as Lanczos iteration
All based on idea that matrix-vector products preferred over matrix-matrix products
Provide only incomplete information about eigenvalues and eigenvectors
Sparsity
- Many matrices that we meet in physical applications are sparse, meaning that most of elements are zero
\[ \left[-\frac{\hbar^2}{2m}\frac{d^2}{dx^2} + V(x)\right]\psi(x) = E\psi(x) \]
\[ \frac{d^2}{dx^2} \sim \frac{1}{\Delta x^2}\begin{pmatrix} -2 & 1 & 0 & 0 & 0 & \cdots & 1 \\ 1 & -2 & 1 & 0 & 0 & \cdots & 0 \\ 0 & 1 & -2 & 1 & 0 & \cdots & 0 \\ \cdots & \cdots & \cdots & \cdots & \cdots & \cdots & \cdots \\ 1 & 0 & 0 & \cdots & 0 & 1 & -2 \end{pmatrix} \]
No point iterating over a whole row to multiply this matrix into a vector representing wavefunction
Only need to store the non-zero values of a matrix (and their locations)
Variety of data structures implemented in
scipy.sparsemoduleMatrix operations from
scipy.sparse.linalguse these structures effciently
Alternative approach: pass matrix operations in
scipy.sparse.linalga function which performs matrix-vector multiplicationInstantiate a
LinearOperatorwith the functionWe’ll see an example shortly
Singular value decomposition
Often faced with need to truncate large matrices in some way due to limits of finite storage space or processing time
What is “right” way to perform truncation?
Singular value decomposition (SVD) is natural in some settings: statistics, signal processing, quantum mechanics…
- SVD is an example of matrix factorization
\[ M = U\Sigma V \]
\(U\) and \(V\) unitary; \(\Sigma\) diagonal with non-negative real entries
SVD completely general: applies to rectangular matrices
If \(M\) is \(m\times n\), \(U\) is \(m\times m\), \(V\) is \(n\times n\), and \(\Sigma\) is \(m\times n\)
\(\min(m,n)\) diagonal elements \(\sigma_i\) of \(\Sigma\) are singular values
Geometrical interpretation
Columns of \(V\) define an orthonormal basis \(\mathbf{v}_i\in \mathbb{C}^n\) (\(i=1,\ldots n\))
\(U\) defines a basis \(\mathbf{u}_i\in \mathbb{C}^m\) \(i=1,\ldots m\)
If we act on \(\mathbf{v}_i\) with \(M\) (to the left) we get \(\sigma_i \mathbf{u}_i\)
Number of nonzero singular values is rank of matrix
Equal to number of independent rows or columns
For general rectangular matrix rank is \(\min(m,n)\)
Often want to produce low rank approximation
Need to define how well the matrix is approximated by the lower rank matrix \(M_r\) of rank \(r<\min(m,n)\)
One possibility: Frobenius norm of \(M-M_r\) should be as small as possible.
Frobenius norm \(\|A\|_{\mathrm{F}}\) of a matrix \(A\) is
\[ \begin{equation} \|A\|_{\mathrm{F}}^2=\sum_i^m \sum_j^n\left|A_{i j}\right|^2 \end{equation} \]
\(\exists\) following simple result: best low rank approximation of rank \(r\) obtained by taking SVD and discarding all but \(r\) largest singular values from matrix \(\Sigma\)
i.e. retain only \(r\) “most important” directions \(\mathbf{v}_i\in \mathbb{C}^n\) and \(\mathbf{u}_i\in \mathbb{C}^m\)
SVD can be computed using
np.linalg.svdTry this fun demo of image compression with SVD
SVD in quantum mechanics
SVD arises naturally in QM of composite systems (with two subsystems)
Example: two spins \(\mathbf{S}_A\) and \(\mathbf{S}_B\)
Hilbert space of each spin has dimension \(n_{A,B}\equiv 2S_{A,B}+1\), where \(\mathbf{S}_{A,B}^2=S_{A,B}(S_{A,B}+1)\) (e.g. 2 for spin-1/2).
General state lives in \(n_A\times n_B\) dimensional Hilbert space
Write in terms of basis vectors \(\ket{a}_A\) and \(\ket{b}_B\) for A and B subsystems as \[ \ket{\Psi_{AB}} = \sum_{a=1}^{n_A}\sum_{b=1}^{n_B} \psi_{ab}\ket{a}_A\ket{b}_B \]
Regard components \(\psi_{ab}\) as a matrix and perform SVD
Equivalent to finding new orthonormal bases \(\ket{\tilde n}_{A,B}\) for two spaces s.t. action of \(\psi_{ab}\) maps between basis vectors of two subsystems (with rescaling)
- In new bases, state \(\ket{\Psi_{AB}}\) is
\[ \ket{\Psi_{AB}} = \sum_{n=1}^{\min(n_A,n_B)} \lambda_n\ket{\tilde n}_A\ket{\tilde n}_B. \]
Note single sum c.f. double sum before
This is Schmidt decomposition: just a restatement of SVD
\[ \ket{\Psi_{AB}} = \sum_{n=1}^{\min(n_A,n_B)} \lambda_n\ket{\tilde n}_A\ket{\tilde n}_B \]
Singular values — or Schmidt coefficients — quantify entanglement of state (2022 Nobel prize)
If only one nonzero singular value state is a product state: no correlations between subsystems
This might not have been evident in original form
- Bell states of two spin-1/2 subsystems
\[ \begin{equation} \left|\Psi^{+}\right\rangle=\frac{1}{\sqrt{2}}\left(|0\rangle_A \otimes|1\rangle_B+|1\rangle_A \otimes|0\rangle_B\right) \end{equation} \]
- Already written in Schmidt form and the two singular values are both \(\frac{1}{\sqrt{2}}\), indicating maximal entanglement
Other applications of SVD
- Applications of SVD in recommender systems, described in this blog post
Many body physics & tensor methods
We saw that state of a quantum system composed of two subsystems represented as a matrix \(\psi_{ab}\)
Generalizes to \(N\) subsystems: wavefunction may a tensor of rank \(N\): \(\psi_{a_1,\ldots a_N}\)
Each index \(a_i\) ranges over dimension of Hilbert space of corresponding subsystem
Penrose tensor notation
Graphical notation due to Roger Penrose
Rank \(N\) tensor is represented as blob with \(N\) legs:
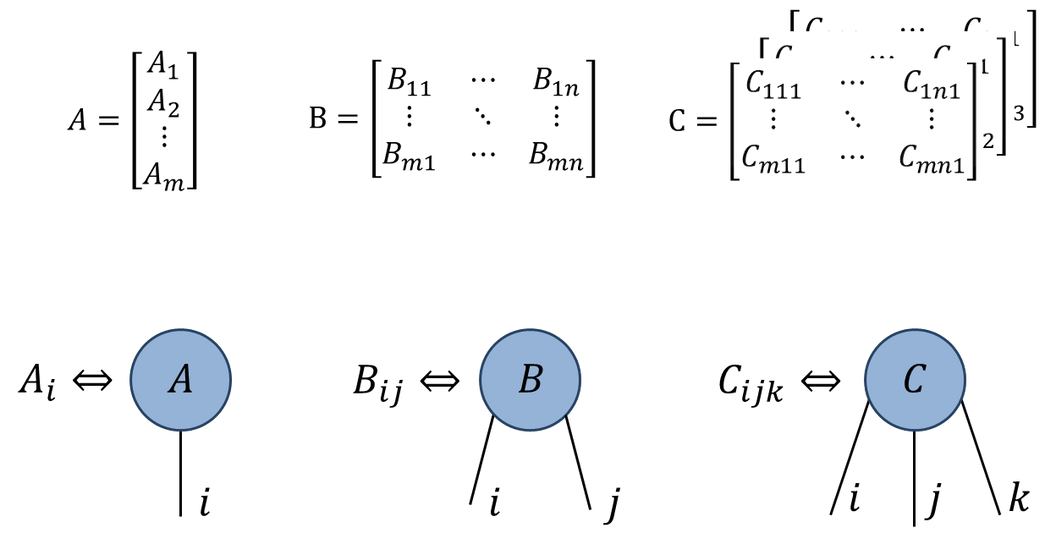
- Represent tensor contractions by connecting legs:
Example: ground state of spin chain
- Simplest example: Heisenberg chain for spin-1/2:
\[ H = \sum_{j=1}^N \left[\sigma^x_j \sigma^x_{j+1} + \sigma^y_j \sigma^y_{j+1} + \sigma^z_j \sigma^z_{j+1} \right] \]
\(\sigma^{x,y,z}\) are usual Pauli matrices and subscript \(j\) means that matrix acts only the \(j\)th index of the wavefunction
Usually impose periodic boundary conditions: \(\sigma^a_{j+N}=\sigma^a_j\)
- In tensor diagram notation
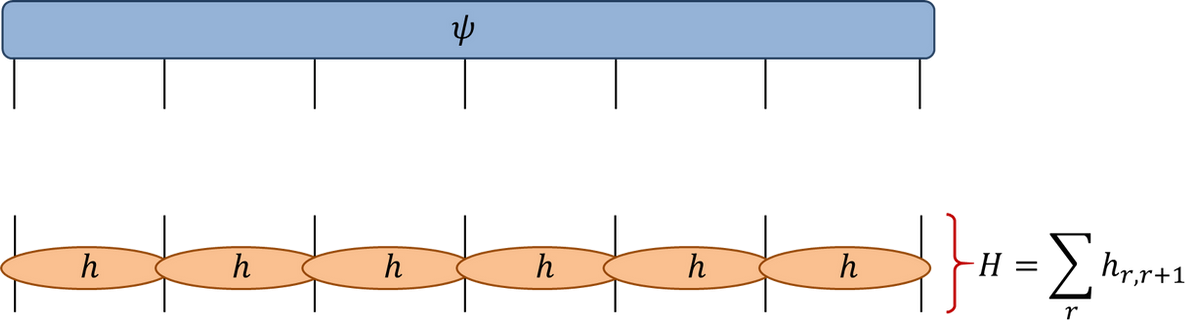
- Number of components of wavefunction \(\psi_{a_1,\ldots a_N}\) is \(2^N\)
\[ H\ket{\Psi} = E\ket{\Psi} \]
Naive matrix-vector multiplication has complexity \(O(2^{2N})\): very bad idea.
Take advantage of structure, using sparsity of Hamiltonian
\(H=\sum_j h_{j,j+1}\) consists of a sum of local terms, each acting on neighbouring pair
- Define function that acts on wavefunction with each \(h_{j,j+1}\)
# by Glen Evenbly (c) for www.tensors.net, (v1.2) - last modified 6/2019
def doApplyHam(psiIn: np.ndarray,
hloc: np.ndarray,
N: int,
usePBC: bool):
"""
Applies local Hamiltonian, given as sum of nearest neighbor terms, to
an input quantum state.
Args:
psiIn: vector of length d**N describing the quantum state.
hloc: array of ndim=4 describing the nearest neighbor coupling.
N: the number of lattice sites.
usePBC: sets whether to include periodic boundary term.
Returns:
np.ndarray: state psi after application of the Hamiltonian.
"""
d = hloc.shape[0]
psiOut = np.zeros(psiIn.size)
for k in range(N - 1):
# apply local Hamiltonian terms to sites [k,k+1]
psiOut += np.tensordot(hloc.reshape(d**2, d**2),
psiIn.reshape(d**k, d**2, d**(N - 2 - k)),
axes=[[1], [1]]).transpose(1, 0, 2).reshape(d**N)
if usePBC:
# apply periodic term
psiOut += np.tensordot(hloc.reshape(d, d, d, d),
psiIn.reshape(d, d**(N - 2), d),
axes=[[2, 3], [2, 0]]
).transpose(1, 2, 0).reshape(d**N)
return psiOutComplexity is \(O(N 2^N)\)
\(2^N\) arises from tensor contractions over indices of a pair of sites for each assignment of the remaining \(N-2\) indices
Still exponential, but exponentially better than \(O(4^N)\)!
- Use this to instantiate a
LinearOperatorwhich is passed into eigenvalue solver (scipy.sparse.linalg.eigsh)
"""
by Glen Evenbly (c) for www.tensors.net, (v1.2) - last modified 06/2020
"""
from scipy.sparse.linalg import LinearOperator, eigsh
from timeit import default_timer as timer
# Simulation parameters
model = 'XX' # select 'XX' model of 'ising' model
Nsites = 18 # number of lattice sites
usePBC = True # use periodic or open boundaries
numval = 1 # number of eigenstates to compute
# Define Hamiltonian (quantum XX model)
d = 2 # local dimension
sX = np.array([[0, 1.0], [1.0, 0]])
sY = np.array([[0, -1.0j], [1.0j, 0]])
sZ = np.array([[1.0, 0], [0, -1.0]])
sI = np.array([[1.0, 0], [0, 1.0]])
if model == 'XX':
hloc = (np.real(np.kron(sX, sX) + np.kron(sY, sY))).reshape(2, 2, 2, 2)
EnExact = -4 / np.sin(np.pi / Nsites) # Note: only for PBC
elif model == 'ising':
hloc = (-np.kron(sX, sX) + 0.5 * np.kron(sZ, sI) + 0.5 * np.kron(sI, sZ)
).reshape(2, 2, 2, 2)
EnExact = -2 / np.sin(np.pi / (2 * Nsites)) # Note: only for PBC
# cast the Hamiltonian 'H' as a linear operator
def doApplyHamClosed(psiIn):
return doApplyHam(psiIn, hloc, Nsites, usePBC)
H = LinearOperator((2**Nsites, 2**Nsites), matvec=doApplyHamClosed)
# do the exact diag
start_time = timer()
Energy, psi = eigsh(H, k=numval, which='SA')
diag_time = timer() - start_time
# check with exact energy
EnErr = Energy[0] - EnExact # should equal to zero
print('NumSites: %d, Time: %1.2f, Energy: %e, EnErr: %e' %
(Nsites, diag_time, Energy[0], EnErr))NumSites: 18, Time: 4.23, Energy: -2.303508e+01, EnErr: 1.065814e-14- Check out Glen Evenbly’s site is you’d like to learn more about these methods!